Full Text (PDF)
Pediatric Education and Research 12(2):p 51-53, July - Dec 2024. | DOI: https://dx.doi.org/10.21088/per.2321.1644.12224.3
Case Report
Apert’s Syndrome: A Rare Case Report
Rajeev Kumar Thapar, , Sumanth Baditela1 , Rajeev Kumar Thapar2 , Meenakshi Bothra3
Author Information
Licence:
Pediatric Education and Research 12(2):p 51-53, July - Dec 2024. | DOI: https://dx.doi.org/10.21088/per.2321.1644.12224.3
How Cite This Article:
Sumanth Baditela, Rajeev Kumar Thapar, Meenakshi Bothra, Apert’s Syndrome: A Rare Case Report. Pediatr. Edu. Res. 2024;12(2): 51-53.
Timeline
Received : N/A
Accepted : N/A
Published : N/A
Abstract
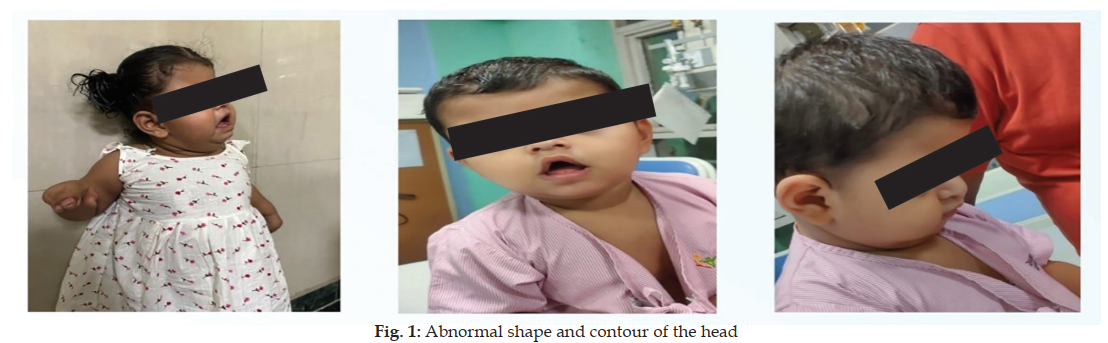
Background: Apert syndrome is a genetic disorder inherited in an autosomal dominant manner, with an occurrence rate of about 15 cases per 100,000 live births. It is caused by a mutation in the fibroblast growth factor receptor-2 (FGFR-2) gene located on chromosome 10q26. The condition mainly impacts the first and second branchial arches, leading to the early closure of cranial sutures (craniosynostosis) with fusion of fingers and toes of the hands and feet. Apert syndrome is rare in India, and a case report is presented. Clinical Description: 21 months female toddler presented with global developmental delay along with distinctive craniofacial features. Clinically toddler exhibited an abnormal head shape and contour, characterized by turribrachycephaly, a depressed nasal bridge, frontal bossing, midface hypoplasia, and a characteristic “crossbow” appearance of the upper lip. Limb examination revealed symmetrical soft tissue syndactyly affecting all digits. Management & Outcome: This case is notable for its rarity and the similarity of its features to other craniosynostosis syndromes, such as Crouzon and Pfeiffer syndromes, posing a diagnostic challenge. Therefore, genetic counselling for the family was recommended, along with early intervention for the child, including plastic surgery for the affected limbs. Conclusion: Acrocephalosyndactyly is an autosomal dominant condition seen rarely, marked by craniosynostosis, craniofacial deformities, and pronounced symmetrical clubbing of fingers and toes of the hands and feet. In the majority of Apert syndrome cases, the condition occurs sporadically, often due to new mutations in the relevant gene.
References
No records found.
Data Sharing Statement
There are no additional data available
Funding
This research received no funding
Author Contributions
All authors contributed significantly to the work and approve its publication
Ethics Declaration
This article does not involve any human or animal subjects, and therefore does not require ethics approval
Acknowledgements
Information Not Provided
Conflicts of Interest
No conflicts of interest in this work
About this article
Cite this article
Sumanth Baditela, Rajeev Kumar Thapar, Meenakshi Bothra, Apert’s Syndrome: A Rare Case Report. Pediatr. Edu. Res. 2024;12(2): 51-53.
Licence:
| Received | Accepted | Published |
|---|---|---|
| N/A | N/A | N/A |
DOI: https://dx.doi.org/10.21088/per.2321.1644.12224.3
Keywords
AcrocephalosyndactylyCraniosynostosisMidface hypoplasia.Search for Similar Articles
Similar Articles
- TB Elimination from India-Challenges and the Path Ahead
- Spontaneous Pneumomediastinum with Subcutaneous Emphysema after a Trivial Injury...
- Disseminated Methicillin-Resistant Staphylococcus Aureus (MRSA) Infection with S...
- Hypospadias with Megameatus Intact Prepuce: A Case Report
- SARS-CoV-2 Seroprevalence among Children
Article Level Metrics
Last UpdatedSaturday 07 February 2026, 16:08:07 (IST)
201
Accesses
0
47
00
Citations
NA
NA
NA
Download citation
Article Keywords
Keyword Highlighting
Highlight selected keywords in the article text.
Timeline
| Received | N/A |
| Accepted | N/A |
| Published | N/A |